Hirschsprung Disease
Hirschsprung disease is a congenital defect in the innervation of the distal intestine, usually limited to the rectum and colon, resulting in partial or total functional obstruction. The symptoms are severe constipation and abdominal distension. Diagnosis is based on barium enema and rectal biopsy. Anal manometry can be useful in the assessment and reveals the abnormal relaxation of the internal anal sphincter. The treatment is surgical.
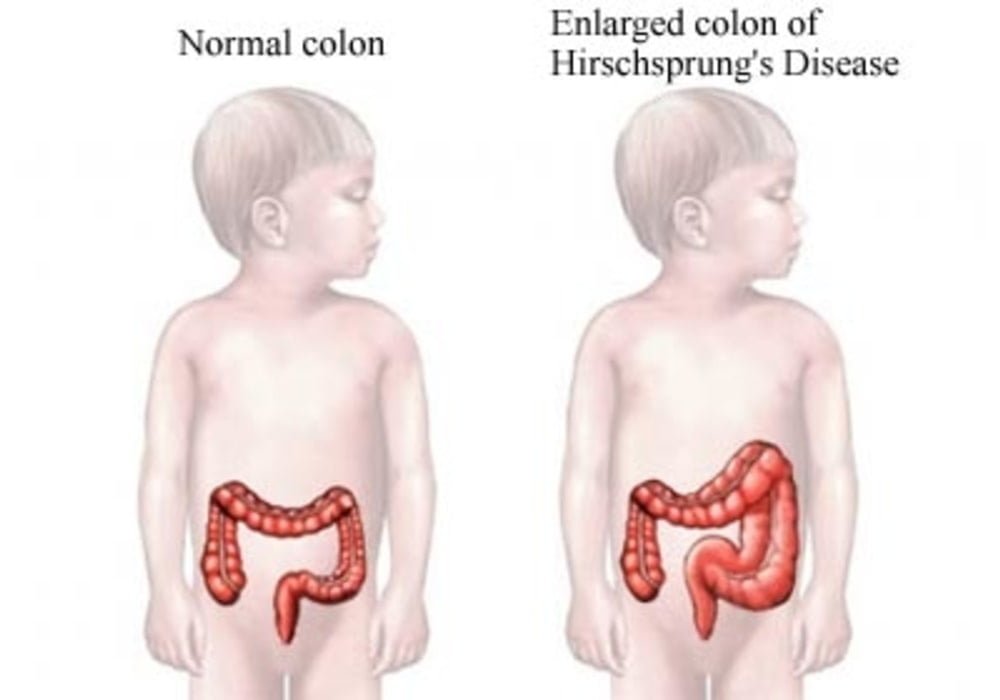
This pathology appears from birth and is the consequence of an absence of nerve ganglia (cells forming a bulge on the path of the nerve) in the wall of the intestine.
The swallowing of food through the digestive tract until it is excreted is, in large part, possible thanks to intestinal peristalsis. This peristalsis is a set of contractions of the intestinal muscles allowing the advance of the food bolus along the digestive tract.
In this situation where there is an absence of nerve ganglia in the large intestine, peristalsis is no longer provided by the body. In this sense, a dilation of the intestine and an increase in its volume is created.
The associated symptoms are all the more important if the area of the nerve ganglia is small.
Causes
It is a polygenic disease, this is due to the mutation of several independent genes, a particular combination of which determines this malformation.
Symptoms of Hirschsprung Disease
Intestinal transit is controlled by the nervous system. Nerve ganglia are therefore located in the intestine allowing the transfer of information from the brain for the control of intestinal peristalsis and thus the progression of food along the digestive tract.
An absence of these nodes, in the case of Hirschsprung’s disease, prevents any transmission of information and thus blocks intestinal peristalsis. Food can no longer pass through the intestines and ends up blocked in the digestive tract.
Symptoms of this disease are usually noticeable very early at birth. However, in some cases, they can appear after one or two years.
Symptoms affecting newborns and children are mainly:
– transit difficulties;
– an inability to expel meconium (first excrements of the newborn) during the first 48 hours;
– constipation;
– jaundice;
– vomitings;
– diarrhea;
– abdominal pain;
– undernourishment.
Symptoms affecting older children are:
– severe constipation with complications (failure to thrive in height and weight);
– poor nutrition;
– abdominal distension;
– a fever.
The child may also develop intestinal infections, such as enterocolitis.
Additional abnormalities may also be visible: sensorineural hearing loss (Waardenburg-Shah syndrome), intellectual disability (Mowat-Wilson syndrome), central alveolar hypoventilation (Haddad syndrome), limb abnormalities (Bardet-Biedl syndrome), medullary thyroid cancer (multiple endocrine neoplasia type 2B) or chromosomal abnormalities (Down syndrome).
The origins of the Hirschsprung Disease
Hirschsprung’s disease is caused by an abnormality in the development of the enteric nervous system. It is an aganglionosis, ie an absence of nerve ganglia (also called “Cajal cells”) in the intestines. This lymph node deficit is more particularly located in the terminal part of the large intestine (colon).
In the subject affected by this pathology, this part of the intestine therefore remains in a state of tonic and permanent contraction. This situation leads to intestinal obstruction.
Both genetic and environmental factors have been implicated in the development of Hirschsprung’s disease.
Indeed, certain genes have been demonstrated in the development of this pathogenesis. It is a polygenetic disease which concerns in particular the genes:
– Ret proco-oncogene (RET);
– the glial cell-derived neutrotrophic factor gene (GDNF);
– the type B endothelin receptor gene (EDNRB);
– the endothelin 3 gene (EDN3);
– the gene for endothelin 1 converting enzyme 1 (ECE1);
– the gene for the cell adhesion molecule L1 (L1CAM).
Name and frequency
The first systematic describer (1886) is the Danish pediatrician Harald Hirschsprung (1830–1916).
On average, this malformation occurs in 1 in 5000 children, with boys being affected significantly more frequently than girls in comparison . Hirschsprung’s disease can be detected in around 12% of infants with Down syndrome (trisomy 21). Combinations with other malformations (e.g. cystic fibrosis , brachydactyly , colon atresia ) occur but are rare.
In 80% of cases, aganglionosis only affects the rectum and / or sigmoid colon (short-segment aganglionosis). About 5% belong to long-segment aganglionosis, in which the pathologically altered section of the large intestine is a total of 40 cm and more. In less than 5% of the cases the nerve cells are missing in the entire section of the large intestine, in this case one speaks of a Jirásek-Zuelzer-Wilson syndrome . In some cases the nerve cells are missing as far as the small intestine.
Risk factors of Hirschsprung Disease
As stated previously, Hirschsprung’s disease is the consequence of the absence of nerve ganglia in the large intestine up to the anus, preventing intestinal peristalsis and therefore the ascending of food to this level.
This deficit of Cajal cells (nerve ganglia) is the consequence of a deficit in the growth of these cells during fetal development. The causes of this lack of cell growth before birth are not yet known. Nevertheless, the possibility of a relationship between the general health of the mother during her pregnancy period and the absence of this type of cells in the fetus has been put forward.
Many genes have been shown in the development of the disease. The presence of these genes can be frequent within the same family. A part of heredity would then be at the origin of the development of this disease.
In addition, certain pathologies can also be an additional risk factor in terms of the development of Hirschsprung’s disease. This is particularly the case with Down syndrome.
Diagnosis
The differential diagnosis is made according to the characteristic symptoms of the disease presented by the subject: intestinal obstruction, anorectal stenosis, pelvic tumors, etc.
The diagnosis most often associated with the disease is made through a rectal biopsy. This biopsy shows the presence or absence of nerve ganglia in the large intestine. In addition, an overexpression of acetylcholine esterase (enzyme allowing acetylcholine to be hydrolyzed into acetic acid and choline).
Prevention and treatment of Hirschsprung’s Disease
Differential diagnosis is made according to the characteristic symptoms of the disease presented by the subject: intestinal obstruction, anorectal stenosis, pelvic tumors, etc.
The diagnosis most often associated with this disease is made by rectal biopsy. This biopsy shows the presence or absence of nerve ganglia in the colon. In addition, overexpression of acetylcholine esterase (an enzyme that allows acetylcholine to be hydrolyzed to acetic acid and choline).
Barium enema (x-ray examination to visualize the colon) can also be performed in the diagnosis of this pathology. This method makes it possible to visualize transient areas devoid of nerve cells, indicating the development of Hischsprung’s disease. However, this diagnostic technique is not 100% reliable. Indeed, 10 to 15% of cases of Hirschsprung’s disease will go undiagnosed after these diagnostic efforts.
The most important treatment for this disease is surgery. This allows ablation of the part of the intestine that lacks nerve cells.
In cases of complete damage to the colon, a colon transplant may be necessary.
After this, an ostomy (a surgical technique that allows to create a connection between two organs) may be performed to connect the operated part of the intestine to the anus or to the upper part of the intestine. This stoma can be permanent or temporary depending on the case.
Surgery helps reduce symptoms associated with the disease. However, the prognosis is incomplete and inflammatory complications can be present and fatal.
Hirschsprung enterocolitis
Hirschsprung enterocolitis is a life-threatening complication of Hirschsprung disease characterized by acute dilation of the colon, often followed by sepsis and shock.
The etiology of Hirschsprung enterocolitis appears to be the significant intestinal dilation upstream of the occlusion, with thinning of the colonic wall, bacterial overgrowth and translocation of intestinal bacteria. Sepsis or shock can develop (more often when the entire colon is affected by Hirschsprung disease) and quickly lead to death; mortality is about 1%. Close monitoring of infants with Hirschsprung disease is therefore essential.
Hirschsprung enterocolitis most often occurs in the first months of life before surgical correction but can also be seen postoperatively, usually in the first year after surgery. Infants who have Down syndrome have an increased risk of developing this complication postoperatively. Infants initially have fever, abdominal distension, diarrhea (which may be hemorrhagic) and subsequently severe constipation.
A barium enema should not be performed in patients with suspected Hirschsprung enterocolitis due to the risk of perforation.
Initial treatment of Hirschsprung enterocolitis is supportive with fluid resuscitation, decompression using a nasogastric tube and rectal tube, and broad-spectrum antibiotics that include anaerobic coverage (eg, combination of ampicillin, gentamicin and metronidazole; meropenem alone; or piperacillin / tazobactam). Some experts recommend saline enemas for infants who are not seriously ill to flush the colon, but these should be done with care so as not to increase intracolic pressure and lead to perforation. Surgery is the radical treatment in infants who have not yet had surgical repair, as well as in infants who have perforation or necrosis of the intestine. For infants who can be stabilized, definitive surgical treatment may be performed instead of the previous standard approach of an ileostomy or bypass colostomy. The previous standard approach should be taken in severe illness.
Information: Cleverly Smart is not a substitute for a doctor. Always consult a doctor to treat your health condition.
Sources: PinterPandai, Naomi E. Butler Tjaden / Science Direct, PLOS One, Cedars-Sinai, National Organization for Rare Disorders
Photo credit: Nucleus Medical Media, Inc. / Western New York Urology Associates