Retinal Cancer
The retina is the transparent, light-sensitive membrane at the back of the eye. Retinal cancers usually start in the choroid, a dense vascular layer that nourishes the retina. The choroid is located between the retina and the sclera (outer, white envelope of the eye). As the vascularization of the retina depends on the choroid, an alteration of the latter by a retinal cancer process can affect vision.
Symptoms
Early detection of melanoma is crucial. The “ABCDE” rule is a helpful guide for identifying suspicious moles or skin lesions:
- Asymmetry: One half of the mole doesn’t match the other.
- Border: Edges are irregular, ragged, or blurred.
- Color: Varied shades of brown, black, or other colors.
- Diameter: Larger than 6mm (about the size of a pencil eraser).
- Evolving: Changes in size, shape, color, or symptoms such as itching or bleeding.
Other warning signs include new growths, sores that don’t heal, or changes in existing moles. Melanoma can develop anywhere on the body, even in areas not exposed to the sun
Retinal cancer and choroidal melanoma are two different types of eye cancer.
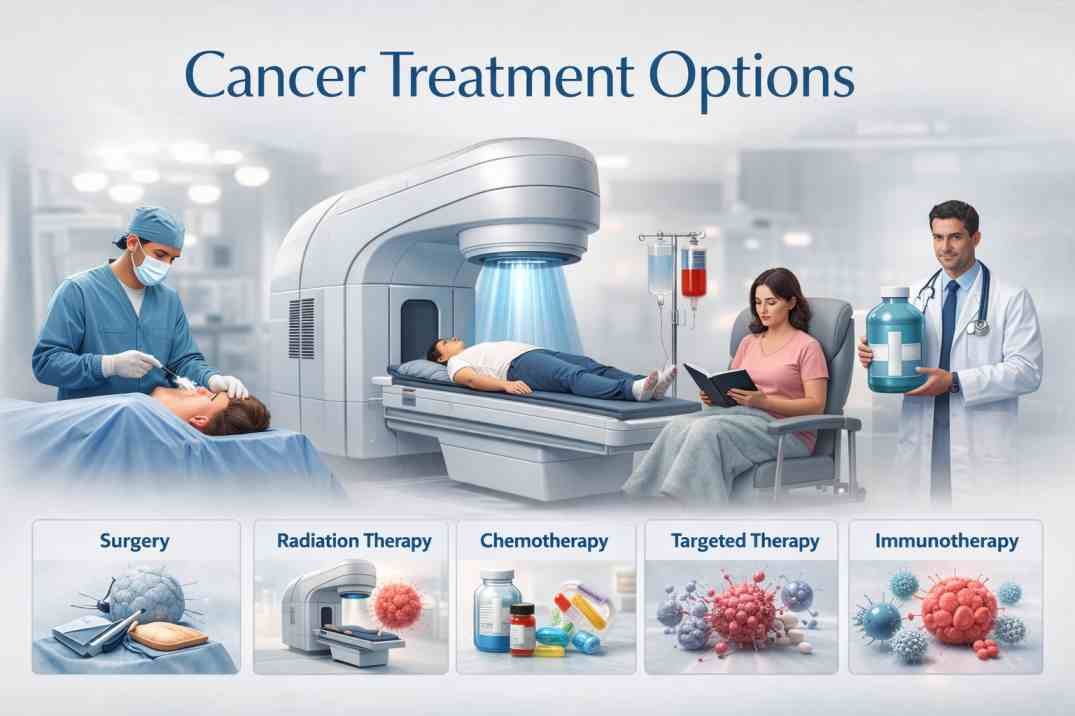
Retinal cancer, also known as retinoblastoma, is a rare cancer that develops in the retina, which is the tissue lining the back of the eye that is responsible for sensing light and sending visual signals to the brain. Retinoblastoma usually affects young children and can be hereditary or non-hereditary. Symptoms may include a white pupil or a noticeable difference in the appearance or color of the eyes. Treatment for retinoblastoma may include surgery, radiation therapy, or chemotherapy.
Choroidal melanoma, on the other hand, is a type of cancer that develops in the choroid, which is the layer of blood vessels that supplies the retina with nutrients and oxygen. Choroidal melanoma is a rare cancer that affects adults and is more common in people with fair skin and light eyes. Symptoms may include blurred vision, a dark spot on the iris or vision loss. Treatment for choroidal melanoma may include surgery, radiation therapy, or a combination of both.
Both retinal cancer and choroidal melanoma are serious conditions that require prompt diagnosis and treatment by an ophthalmologist or a specialist in eye cancer. Early detection is important for a better outcome and preservation of vision.
Choroidal melanoma
Choroidal melanoma is a cancer that originates from cells that produce choroidal pigment (melanocytes).
It is the most common eye cancer. It is more common in people with white skin. It occurs most frequently between the ages of 55 and 60.
Symptoms
At the initial stage, visual acuity is most often normal. Later, it can blur vision or cause retinal detachment, with symptoms like flashes, or a veil or curtain across the visual field, or a sudden increase or change in floaters (objects that appear to move at across the field of vision). Melanomas, especially large ones, can spread into the eye socket or spread through blood (metastasis) to other parts of the body and can be fatal.
Diagnostic
Clinical eye examination
Other tests
Early diagnosis of choroidal melanoma is important because smaller tumors are easier to treat.
Diagnosis is made with an ophthalmoscope and tests including ultrasound, fluorescein angiography, and serial photography.
Treatment
For small tumours, laser, radiotherapy or implant
For large tumours, removal of the eye
If the melanoma is small, laser treatment, radiation, or injection of a radioactive substance can preserve visual acuity and save the eye.
When the cancer is large, the eye must be removed.
Choroidal metastases
Choroidal metastases are cancerous tumors originating from other parts of the body and spreading to the eye. Because of its rich vascularization, the choroid is often the site of metastases from other organs. In women, the main cause is breast cancer. In men, lung and prostate cancer are more frequently involved.
Symptoms
Often, choroidal metastases do not cause symptoms when they are not in the advanced stage. Symptoms, when they do appear, are often loss of visual acuity or symptoms of retinal detachment such as floaters, flashes of light, blurred vision and haze in the field of vision. Vision loss can vary from mild to severe.
Diagnostic
Clinical eye examination
- Usually ultrasound
- Biopsy
- Sometimes the diagnosis of choroidal metastases is made by ophthalmoscope during a routine ophthalmological examination. The diagnosis is confirmed by ultrasound.
Confirmation of the diagnosis can be made by removing, with a fine needle, a small piece of the tumor which will be examined under a microscope (biopsy).
Treatment
Chemotherapy and/or radiotherapy
Chemotherapy or radiotherapy, or both at the same time, are the usual treatments for this type of choroidal metastases and depend on the type of primary cancer.
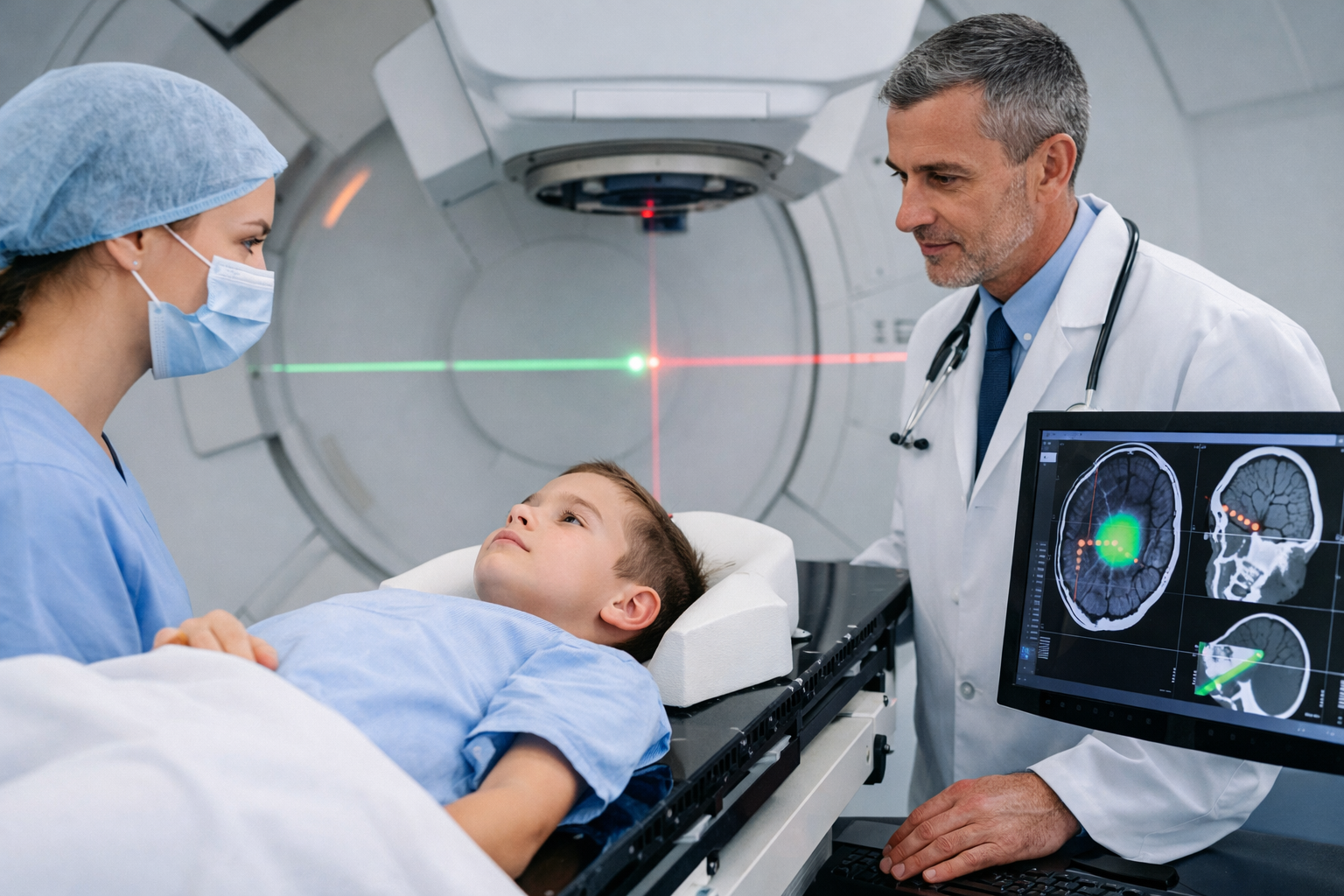
Retinoblastoma
Retinoblastoma is a malignant tumor of the retina, rare (one child in 15 to 20,000 would be affected) and of genetic origin, usually appearing before the age of 5 years.
Symptoms
Two known signs indicate a risk of retinoblastoma in a person:
– leukocoria (whitish reflection of the pupil, appearing – at a certain angle, and sometimes highlighted in photos taken with a flash)
– a strabismus.
The diagnosis of retinoblastoma is made by an ophthalmologist. This carries out a fundus examination with dilation of the pupil and, in the event of suspicion of retinoblastoma, refers the child to a specialized center which will carry out a new dilated fundus examination and an examination of the retina, the more often under general anesthesia.
The ophthalmological examination can be supplemented by MRI and ocular ultrasound. In the event of a very large tumour, retinal or optic nerve invasion, the search for distant metastases may be necessary.
Treatment
The management of retinoblastoma has been the subject of recommendations published by the Canadian Retinoblastoma Society.
The treatments are either surgery with sometimes surgical removal of the eye, chemotherapy (cisplatin, etoposide and vincristine), diode laser, thermo-chemotherapy, cryotherapy, radiotherapy or photocoagulation.
Enucleation is often the only option in non-diffuse forms in developing countries. The anatomopathologic examination of the eye makes it possible to see whether the tumor has remained intraocular.
Information: Cleverly Smart is not a substitute for a doctor. Always consult a doctor to treat your health condition.
Sources: PinterPandai, Harvard University, Associated Retina Consultants
Main photo credit: Morleyj / Wikimedia Commons (Public Domain) Author: Morleyj
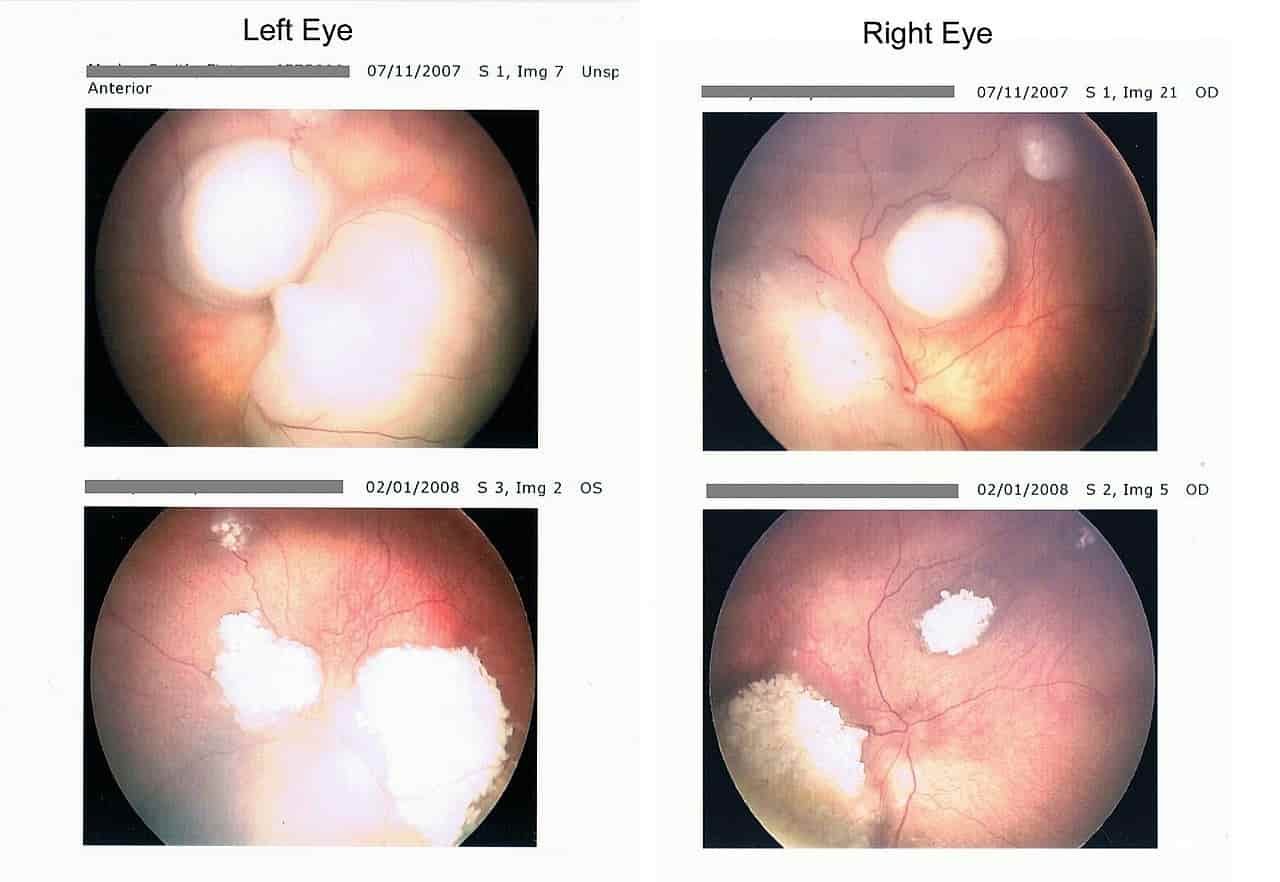
Main photo description: Retinoblastoma (retinal cancer) retina scan before and after chemotherapy.